Le Diabète sucré
Complications
Retour
Le diabète, quil soit insulino-dépendant
(DID) ou non (DNID) est susceptible de provoquer des complications à
moyen et long terme. Lapparition de ces complications est fonction de
léquilibration du diabète. Un diabète équilibré
se compliquera moins vite quun diabète livré à lui-même.
Lorsquil nest pas traité, le DID évolue invariablement
vers un coma acido-cétosique et la mort. Mais malgré le traitement
à base dinsuline les complications restent fréquentes et
nombreuses. Le diabète favorise les infections, en particulier la
tuberculose, les mycoses cutanées et génitales (témoins
dun mauvais équilibre du diabète), des infections cutanées,
les maladies virales et des infections urinaires.
Mais outre les infections fréquentes et diverses, on peut distinguer
trois grands types de complications (fig.1) :
-
La macroangiopathie, touchant les artères, responsable dhypertension
artérielle (HTA), daccidents cardio-vasculaires et vasculaires
cérébraux.
-
Les microangiopathies, cest-à-dire les petites lésions
vasculaires. Elles touchent les artérioles et les capillaires avec
pour cibles privilégiées la rétine et le rein.
-
La neuropathie, ou dégénérescence des petits
nerfs.
Nous étudierons donc la physiopathologie de lAcidocétose
diabétique, de la microangiopathie, de la macroangiopathie
en
insistant sur limplication de lHTA et de la dyslipoprotéinémie
dans la survenue daccidents cardio-vasculaires chez le diabétique.
Enfin nous verrons comment les diverses complications chroniques : macroangiopathie,
microangiopathie, neuropathie et infections peuvent elles-mêmes entraîner
une complication bien connue chez le diabétique : Le Pied Diabétique.
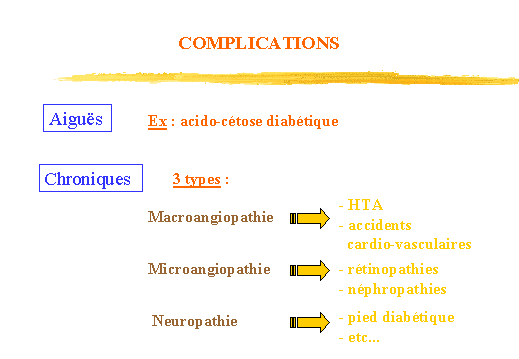
Figure 1
I - Acidocétose diabétique
PHYSIOPATHOLOGIE
Avant linsulinothérapie, lacidocétose aboutissait à
un coma entraînant en quelques heures le décès. Avec
les progrès actuels et léducation des diabétiques,
le terme de coma a pris un sens plus large désignant une acidocétose
sévère dont lincidence annuelle est denviron 5 pour mille.
Lacidocétose, cest à dire une accumulation de corps
cétoniques dans lorganisme est la conséquence dune carence
profonde en insuline. Mais dautres hormones agissent en synergie et jouent
un rôle important dans la cétogénèse par leur
action lipolytique. Ce sont : Le Glucagon, le Cortisol et les Catécholamines.
-
Rôle de la carence en
insuline. (fig. 2)
La chute de linsulinémie lors du jeûne entraîne
la mise en route de la voie catabolique permettant à lorganisme
de puiser dans ses réserves. La lipolyse adipocytaire libère
des AG dont le métabolisme hépatique produit des corps
cétoniques utilisés par le muscle cardiaque. Un autre
mécanisme : la néoglucogénèse hépatique
fournit du Glucose aux tissus gluco-dépendants, principalement le
cerveau dont les besoins quotidiens en glucose sont deux fois supérieurs
aux réserves en glycogène hépatique.
Il existe un rétrocontrôle du catabolisme : la diminution
de linsulino-sécrétion (par exemple lors du jeûne)
active la lipolyse et donc comme nous venons de le voir une augmentation
de la cétogenèse. Or, les corps cétoniques entraînent
une sécrétion dinsuline qui freine en retour la lipolyse
et donc la cétogenèse. Ainsi, chez le diabétique insulinoprive,
labsence de ce rétrocontrôle entraîne des taux dAG
libres 2 à 4 fois supérieurs à la normale (lors du
jeûne).
Donc la carence en insuline va entraîner une hyperglycémie
et une hypercétonémie.
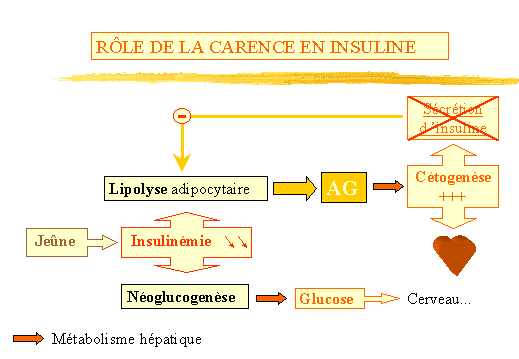 Figure 2
Figure 2
Lhyperglycémie est due :
-
A la diminution de la pénétration intracellulaire du Glucose
en labsence de transport insulino-sensible.
-
A la glycogénolyse hépatique.
-
A lhyperproduction endogène de Glucose : Néoglucogenèse.
Conséquences : (fig. 3) Le glucose ne pénétrant
plus dans la cellule entraîne une hyper-osmolarité extra-cellulaire
induisant un passage deau et de potassium intracellulaire vers le compartiment
extra-cellulaire. Il en résulte une hypervolémie qui va entraîner
une augmentation du flux de filtration glomérulaire. La quantité
de Glucose filtré dépasse la capacité de réabsorption
du tubule rénal et le glucose non réabsorbé est éliminé
dans les urines : glycosurie. Celle-ci saccompagne dune diurèse
osmotique qui non compensée par les boissons entraîne
une hypovolémie qui par opposition à lhypervolémie
va induire une chute du flux glomérulaire appelée insuffisance
rénale fonctionnelle majorant lhyperglycémie.
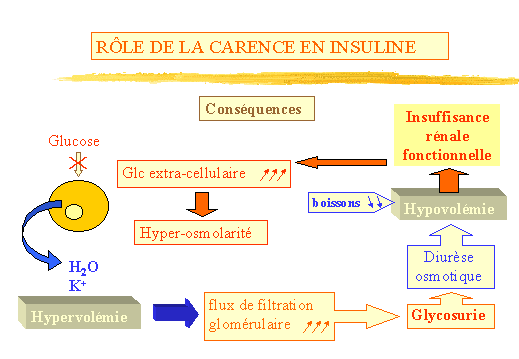
Figure 3
Lhypercétonémie est due (fig. 4) :
-
A une hypercétogenèse. En effet, linsuline est la seule
hormone anti-lipolytique, inhibant la lipase adipocytaire qui est nous
lavons vu responsable de la fourniture dAG pour le métabolisme
hépatique. La carence en insuline accroît donc la lipolyse.
Au niveau hépatique, la b oxydation de
ces AG, produit de lacétyl CoA dont la voie préférentielle
dutilisation est alors la cétogenèse. Loxydation intramitochondriale
est sous la dépendance de l ACT (acyl-carnitine-transférase)
qui permet le passage de lAcyl CoA du cytoplasme à la matrice intramitochondriale.
Or lactivité de cette enzyme dépend du taux de malonyl COA
reflet de lorientation métabolique du foie. Rappelons que nous
sommes dans le cas dune carence en insuline, donc en situation catabolique.
Ainsi la diminution de la concentration en malonyl CoA (substrat utilisé
dans la lipogenèse) stimule lactivité de lACT, et donc
lapport dacétyl CoA nécessaire à la formation des
corps cétoniques.
-
A la diminution de lutilisation des corps cétoniques par les tissus
périphériques en labsence dinsuline.
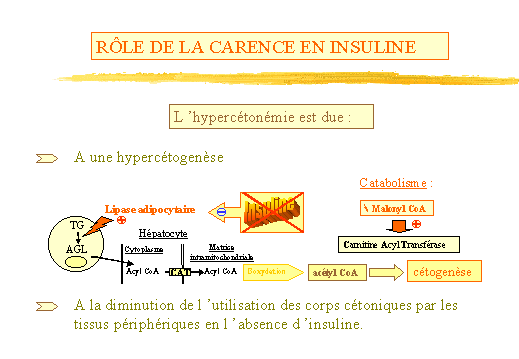
Figure 4
Conséquences :
-
Lhyperproduction dacides cétoniques (acide acéto-acétique
et bêta-hydroxybutirique) qui sont des acides forts donc totalement
ionisés au pH plasmatique entraîne une acidose métabolique
lorsque
les systèmes tampon ne suffisent pas à neutraliser les ions
H+
-
Lélimination des corps cétoniques dans les urines se fait
sous forme de sel de sodium et de potassium entraînant une perte
de ces deux cations avec en échange une réabsorption danions
chlore. Mais nous avons vu quune des conséquences de lhyperglycémie
était une hypovolémie consécutive à la diurèse
osmotique et provoquant une insuffisance rénale fonctionnelle qui
va freiner lélimination des cations.
-
Lhyperventilation avec élimination pulmonaire grâce au système
tampon bicarbonate-acide carbonique qui permet de neutraliser un acide
fort en le transformant en acide faible volatile. La polypnée est
un signe fondamental présent dans 90 à 100 % des cas. Par
ailleurs, on note lodeur caractéristique dacétone exhalée.
(fig. 8)
-
Lacidose grave peut provoquer une dépression respiratoire. Elle
est responsable dune diminution de la contractilité myocardique,
et dune diminution du tonus vasculaire entraînant un collapsus cardiovasculaire.
-
Inhibition de lexcrétion rénale de lacide urique responsable
dune hyper-uricémie.
Ces conséquences que nous venons de voir entraînent leurs
propres conséquences.
Un signe clinique important du coma acidocétosique est la
déshydratation des secteurs intra et extra-cellulaire.
(fig. 5)
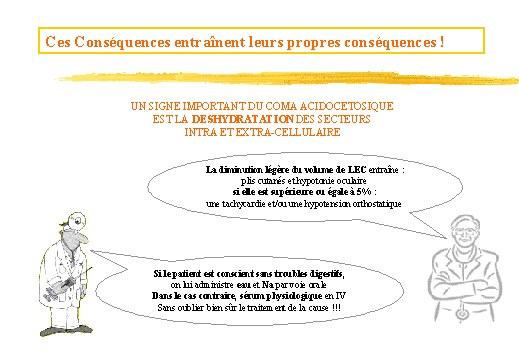
Figure 5
La déshydratation est due :
-
A la diurèse osmotique
-
A la polypnée (élimination dacétone au niveau pulmonaire)
-
Aux vomissements fréquents.
Cette déshydratation entraîne nous lavons dit une hypovolémie
responsable dune insuffisance rénale fonctionnelle avec hyperaldostéronisme
secondaire visant à la réabsorption deau et de sodium au
niveau du tubule rénal. Néanmoins le bilan sodé reste
négatif.
Les pertes de sodium sont dues :
-
A lélimination de corps cétoniques dans les urines sous
forme de sels
-
A la diurèse osmotique
-
Aux vomissements
Les pertes de potassium ont lieu à deux niveaux (fig.
6) :
-
Passage de potassium intra-cellulaire vers le compartiment extra-cellulaire
du :
-
Au catabolisme : glycogénolyse et protéolyse
-
A lhyperosmolarité extra-cellulaire.
-
Elimination du potassium dans les urines due :
-
A lélimination de corps cétoniques dans les urines sous
forme de sels
-
A la diurèse osmotique
-
A lhyperaldostéronisme
Ainsi on remarque que la kaliémie peut être haute normale
ou basse avec malgré tout un bilan négatif.
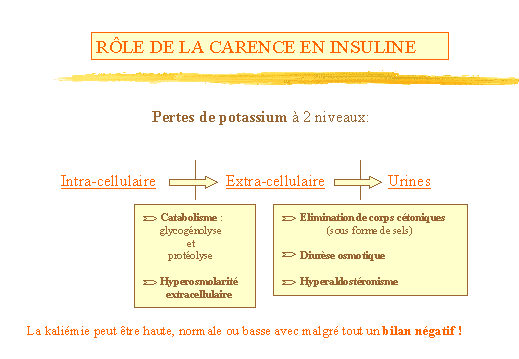
Figure 6
Enfin, lhypophosphatémie (perte du phosphate) entraîne
un déficit en 2,3-diphosphoglycérate présent dans
les hématies, qui favorise la dissociation de loxyhémoglobine
au niveau tissulaire et dont le déficit peut être responsable
dune hypoxie tissulaire avec ses propres conséquences.
-
Hormones de " contre-régulation
" (fig. 7)
Leur action lipolytique ne se manifeste que sil existe une carence en
insuline.
-
Le Glucagon agit à trois niveaux sur le métabolisme
hépatique :
-
Activation de la glycogénolyse et inhibition de la synthèse
du glycogène.
-
Blocage de la glycolyse et activation de la néoglucogenèse.
-
Activation de lACT directement et indirectement par lintermédiaire
dune diminution du taux de malonyl CoA. Il active ainsi la cétogénèse.
Cest le rapport Insuline/Glucagon qui joue un rôle important sur
le métabolisme : il y a anabolisme sil augmente et catabolisme
quand il est diminué.
-
Le Cortisol a une action lipolytique et hyperglycémiante
par augmentation des AA précurseurs de la néoglucogenèse,
induction des enzymes impliquées dans celle-ci et inhibition de
lutilisation périphérique du glucose.
-
Les catécholamines ont une action hyperglycémiante
et lipolytique. Elles stimulent la cétogénèse et inhibent
la sécrétion dinsuline.
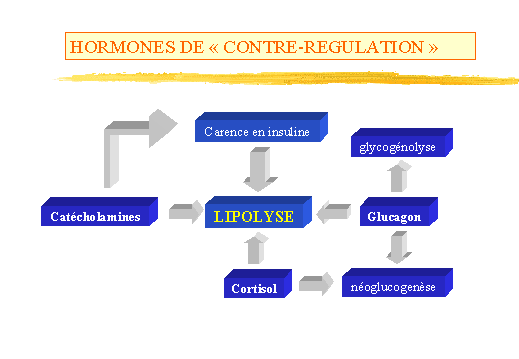
Figure 7
CAUSES
Dans 85% des cas, le coma acidocétosique complique un diabète
traité par linsuline. Mais dans 15% des cas, il complique un diabète
non traité par linsuline soit parce quil sagit dun diabète
auquel vient se surajouter une agression (infection) soit parce quil sagit
dun diabète de type II devenant insulino-dépendant après
des années de traitement par hypoglycémiants oraux.
Ainsi les causes de lacidocétose diabétique sont diverses
:
-
Coma acidocétosique révélateur du diabète
-
Infection
-
Accident cardio-vasculaire (1 fois sur 2 il sagit dune complication due
au diabète). Comme dans le cas dune infection il y a alors élévation
du taux de sécrétion du Glucagon, du Cortisol et des Catécholamines
induisant une insulino-résistance et une diminution de la sécrétion
dinsuline.
-
Arrêt de linsulinothérapie (volontaire ou consécutive
à une panne de pompe à perfusion sous cutanée dinsuline
surtout pour les premières générations de pompes.)
-
Grossesse : les besoins en insuline augmentent dès le deuxième
trimestre de grossesse. Le coma acidocétosique chez la mère
peut entraîner la mort in utero du ftus.
CONCLUSION : Education du diabétique, efficacité des
traitements et rapidité du diagnostique permettent aujourdhui lobtention
dun taux de mortalité inférieur à 5%

Figure 8
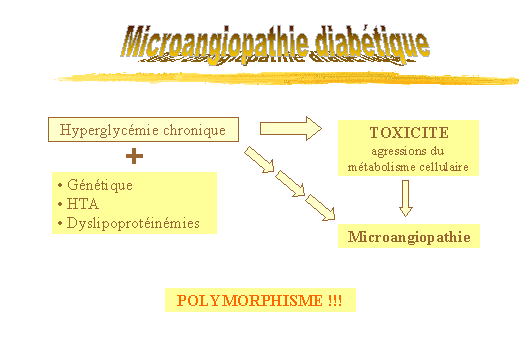
Figure 9
II - La microangiopathie diabétique
PHYSIOPATHOLOGIE
La microangiopathie diabétique intéresse les petits vaisseaux
: artérioles, veinules et capillaires. Elle se manifeste cliniquement
au niveau de lil (rétinopathie), du rein (glomérulopathie)
et des nerfs (neuropathie).
1.Responsabilité de lhyperglycémie
chronique. (fig. 9)
La théorie retenue actuellement sur le déterminisme de
la microangiopathie diabétique est une théorie métabolique
reposant sur la toxicité du glucose et ses mécanismes. Lhyperglycémie
agirait comme une répétition dagressions aiguës du
métabolisme cellulaire. Ce processus semble être sous linfluence
dun déterminisme génétique et régulé
par des facteurs daggravation tels que lHTA et les dyslipoprotéinémies.
Néanmoins, lhyperglycémie joue un rôle nécessaire
mais non suffisant dans la survenue de ces lésions. Les chaînons
sont multiples entre lhyperglycémie et la microangiopathie laissant
envisager lexistence de polymorphismes dans la possibilité dapparition
de complications dégénératives.
2. Mécanismes de la glucotoxicité.
Les mécanismes de la toxicité du glucose au niveau des
tissus cibles des complications du diabète sont multiples. Le glucose
exerce son effet toxique par différentes voies : la voie des polyols,
la déplétion en myoinositol, la glycation protéique,
un défaut en héparane sulfate, le stress oxydatif, la voie
de la protéine kinase C et une action sur lexpression génique.
-
La voie des Polyols (fig. 10)
En présence d'une hyperglycémie, un détournement du
métabolisme du glucose se produit, celui-ci au lieu d'être
essentiellement oxydé dans la voie de la glycolyse se trouve l'être
dans la voie des polyols. Lexcès de glucose qui franchit la membrane
cellulaire est réduit en sorbitol sous laction de laldose
réductase en présence de NADPH, H+ puis la sorbitol-deshydrogénase
catalyse sa transformation en fructose. Compte tenu du Km élevé
de ces enzymes, seule la disponibilité en substrats constitue un
facteur limitant. Laccumulation du sorbitol saccompagne dune déplétion
en myoinositol. En effet, lhyperglycémie va inhiber la captation
du myoinositol par un effet compétitif du glucose sur les récepteurs
membranaires du myoinositol et laccumulation intracellulaire du sorbitol
favorise sa sortie extra-cellulaire. Le défaut en myoinositol entrave
le métabolisme des phospho-inositides, la production de diacylglycérol
et dinositol triphosphate. DAG et inositol triphosphate régulent
lactivité de la protéine kinase C (PKC) et leur déplétion
induit un défaut dactivation de la PKC avec finalement une diminution
de la Na-K ATPase. Celle-ci serait à lorigine dune rétention
intracellulaire du sodium et surtout dun défaut de captation sodium-dépendante
de différents substrats : AA et myoinositol lui-même créant
un véritable cercle vicieux.
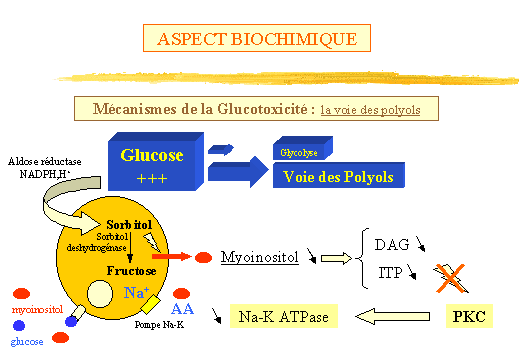
Figure 10
Le défaut dextrusion cellulaire du sodium et les altérations
des échanges membranaires qui lui sont liés expliqueraient
entre autres :
-
Lhyperhydratation du cristallin conduisant à la cataracte.
-
Laltération de lépithélium cornéen responsable
dune kératopathie diabétique.
-
Lhyperperméabilité capillaire et laltération de
lépithélium pigmentaire rétinien responsable de la
rupture de la barrière hémato-rétinienne.
-
Laltération du mésangium glomérulaire.
-
La glycation protéique (fig. 11)
Une des conséquences essentielles de l'hyperglycémie est
la glycosylation non enzymatique ou glycation des protéines. Ce
processus se déroule selon trois étapes :
-
Formation d'une base de Schiff par combinaison de la fonction aldéhyde
du glucose avec les résidus aminés de la protéine,
principalement la lysine et la fonction amine N-terminale, le taux de formation
étant égal au taux de dissociation.
-
Réarrangement d'Amadori, atteignant un équilibre après
quelques semaines, la constante de dissociation ne représentant
que 1.6 % de la constante de formation (réaction quasi-irréversible)
; ces deux étapes aboutissent aux produits de glycation dits précoces
et caractérisent les protéines de demi-vie brèves
ou intermédiaires. (exemple de lhémoglobine)
-
Accumulation lente et irréversible, par réarrangements, transferts
d'hydrogène et formation d'intermédiaires très réactifs,
de produits terminaux de glycation ou produits de Maillard, caractérisant
les protéines structurales de durée de vie prolongée,
et dont les traits biochimiques principaux sont leur pigmentation brune,
leur fluorescence, et leur implication dans la formation de liaisons croisées
entre protéines.
La glycation est ainsi susceptible de modifier les propriétés
de différents enzymes et protéines en fonction de leur demi-vie.
(augmentation de laffinité de lhémoglobine glyquée
pour loxygène ce qui en théorie favorise lhypoxie tissulaire
mais dont les effets restent minimes in vivo)
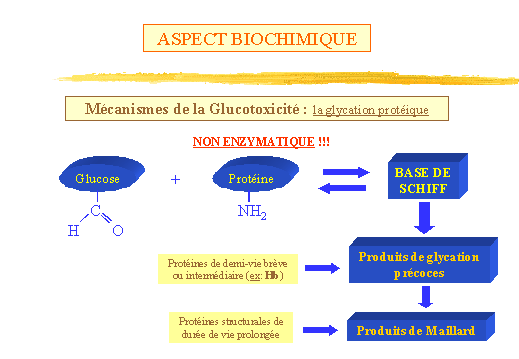
Figure 11
En revanche, les protéines à durée de vie longue
(collagène, myéline) vont subir des réactions complexes,
notamment la réaction de Maillard. Leur accumulation tient de lirréversibilité
de la réaction. Ils seraient responsables :
-
De liaison croisée entre les fibrines du collagène
qui devenant rigide et moins dégradable saccumule au niveau des
membranes basales et du mésangium glomérulaire.
-
De modifications structurales au niveau des membranes basales qui perdent
leur protéoglycanes suite au masquage de leurs sites de liaison
(en particulier lhéparane sulfate) et dont les pores sont élargis.
Lassemblage plurimoléculaire de la membrane basale est perturbé
par la glycation, fragilisant cette dernière et réduisant
ladhésion des cellules endothéliales. Laltération
de linterface endothélium-membrane basale saccompagne dune baisse
des charges négatives par perte de protéoglycanes entraînant
une hyperperméabilité vasculaire.
-
Dune fixation anormale de lalbumine, des globulines et des lipoprotéines
dans la matrice extra-cellulaire de la paroi vasculaire au niveau de groupements
réactifs générés par la glycation.
-
Dune réduction de la susceptibilité à la protéolyse
du collagène et de la membrane basale contribuant à lirréversibilité
de lépaississement des basales.
-
Dune production par les macrophages de cytokines suite à la liaison
de produits terminaux de glycation sur leurs récepteurs macrophagiques,
entraînant notamment une synthèse accrue du collagène
et une prolifération des cellules endothéliales et musculaires
lisses.
En effet, le métabolisme des protéines de la matrice
extra-cellulaire fait intervenir les macrophages, au niveau desquels un
récepteur spécifique des produits terminaux de glycation
a été récemment mis en évidence. Ce récepteur
est inhibé par linsuline et activé par le TNF (tumor necrosis
factor).
La stimulation de ce récepteur induit la sécrétion
de cytokines (TNF et IL-1) et de facteurs de croissance. Cette sécrétion
a pour but le recrutement de cellules mésenchymateuses et endothéliales
qui vont sécréter des collagénases, protéases
et facteurs de croissance pour le remodelage de la matrice extra-cellulaire.
La liaison sur des récepteurs spécifiques de produits terminaux
de glycation va entraîner la prolifération de cellules mésangiales,
endothéliales et fibres musculaires lisses, ainsi quune augmentation
du collagène IV et enfin le catabolisme accru des protéoglycanes
induisant comme nous lavons vu une hyperperméabilité vasculaire
par diminution des charges négatives de la paroi.
N.B. : Lhémoglobine glyquée est un marqueur de léquilibre
glycémique.
-
Défaut en héparane sulfate
Lhyperglycémie chronique entraîne un déficit
relatif en héparane sulfate. En effet ce déficit dépend
dun facteur génétique qui explique la probabilité
variable dun individu diabétique à un autre de développer
une microangiopathie.
Ce déficit expliquerait :
-
Lhyper-perméabilité capillaire.
-
La tendance thrombogène.
-
La stimulation de la prolifération cellulaire.
N.B. : Lhéparine est susceptible de ralentir lévolution
de certaines formes dinsuffisance rénale chronique et inhibe in
vitro la prolifération des cellules endothéliales et mésangiales.
-
Stress oxydatif : excès de radicaux libres (fig. 12-13)
Le diabète est responsable du stress oxydatif à deux
niveaux :
-
Une augmentation des radicaux libres (anion superoxyde, peroxyde dhydrogène,
radical hydroxyle) parallèle au déséquilibre glycémique.
(fig. 12)
-
Une diminution des anti-oxydants endogènes utilisés pour
leur dégradation : formes réduites de la Vitamine C, de la
Vitamine E et Glutathion réduit. (fig. 13)
Causes : On attribue laugmentation de radicaux libres à
:
-
La glycation des protéines qui favorise leur oxydabilité,
les protéines glyquées pouvant réagir avec l'oxygène
pour former des radicaux libres oxygénés. Glucose et protéines
se combinent pour former une base de Schiff que la réaction dAmadori
transforme en produits de glycation " précoces ". La glycation précède
donc loxydation qui aboutit aux produits terminaux de glycation appelés
produits de " Glycoxydation ". (ex : la fructoselysine ou la fructosehydroxylysine
oxydées en carboxymethyllysine et carboxymethylhydroxylysine)
-
L " auto-oxydation " des aldoses (glucose) en composés dicarbonyl
très réactifs (glucosone) capables de se combiner avec des
protéines pour former des composés kétoimine évoluant
vers le brunissement avec production de radicaux libres, cette voie étant
désignée sous le terme de " glycosylation auto-oxydative
".
La déplétion des anti-oxydants est quant à elle attribuée
à :
-
La voie des Polyols, cause essentielle de ce défaut, avec une chute
de NADPH, H+ (consommée par lAldose réductase).
-
La similitude structurale entre glucose et acide ascorbique.
-
La déplétion en myoinositol, entraînant une diminution
de la réabsorption rénale Na-dépendante de lacide
ascorbique.
Ceci induisant naturellement un déficit en Vitamine C.
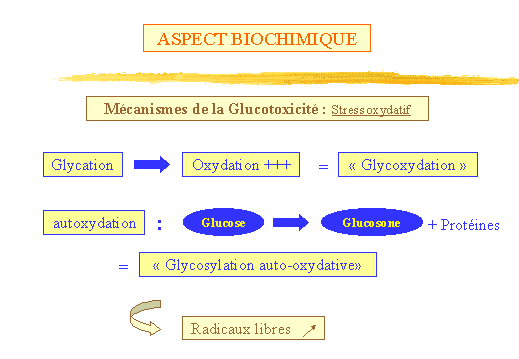
Figure 12
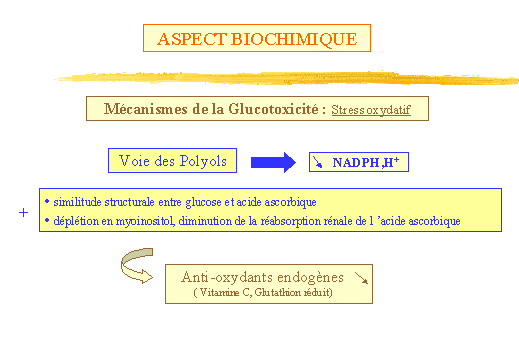
Figure 13
Conséquences : Les radicaux libres provoquent :
-
Une oxydation en chaîne des lipides membranaires entraînant
une désorganisation profonde de la structure des membranes qui deviennent
hyper-perméables.
-
Une accélération de la réaction de Maillard des protéines
glyquées, en particulier des protéines du tissu conjonctif,
formant des produits " glyco-oxydés ".
-
Une inhibition de la production de prostacycline et une augmentation de
celle de thromboxane A2 par les plaquettes.
Il sagit dun véritable cercle vicieux puisque les lésions
tissulaires et la mort cellulaire provoquées par le stress oxydatif
entraînent à leur tour une augmentation des radicaux libres.
-
Activation de la protéine kinase C
La PKC joue un rôle important dans la transduction du signal
apporté par les hormones et les facteurs de croissance. Elle intervient
ainsi dans des fonctions aussi diverses que la croissance cellulaire et
la synthèse d'ADN, le renouvellement des récepteurs membranaires
et la réactivité aux facteurs de croissance, la contraction
des fibres musculaires lisses et le contrôle du tonus vasculaire,
la production de collagène et la synthèse de la membrane
basale, langiogenèse. Son activité est régulée
par le niveau de diacylglycérol (DAG) et d'inositol triphosphate.
Lhyperglycémie chronique augmenterait lactivité de la PKC.
Cet effet est lié à une augmentation de la synthèse
de novo de DAG à partir de précurseurs dérivés
de la glycolyse (fourniture de dihydroxyacetone-phosphate donnant le Glycerol-3-phosphate,
puis un acide phosphatidique et enfin le DAG). Cet effet du glucose sur
la PKC a été retrouvé sur des cellules musculaires
lisses aortiques bovines et au niveau de glomérules rénaux
de rats diabétiques.
Lhyperactivité de la PKC pourrait donc jouer un rôle clé
dans la pathogénie de la microangiopathie diabétique, responsable
dune hyperperméabilité capillaire et dun hyperdébit
(vasodilatation) que lon retrouve dans la rétinopathie. Mais cette
théorie reste contestée : notons quelle est en contradiction
avec la théorie de la déplétion en myoinositol.
-
Action sur lexpression génique
Lhyperglycémie chronique pourrait entraîner une amplification
de lexpression de certains gênes avec une production accrue de collagène
IV, de fibronectine. Il pourrait en être de même pour laldose
réductase (Cf voie des polyols).
Une caractéristique remarquable est l'existence d'une "mémoire
de l'hyperglycémie" définie comme la persistance de la surexpression
génique induite par le glucose après plusieurs divisions
cellulaires. Il est bien établi que les lésions microvasculaires
induites par l'hyperglycémie prolongée sont difficilement
réversibles avec la normalisation glycémique qui doit être
la plus précoce possible. Le mécanisme de cette altération
de lexpression génique reste incertain. Il pourrait y avoir glycation
de lADN.
Une autre hypothèse repose sur une interaction réciproque
cellule endothéliale-matrice extracellulaire, la glycation des composants
de la matrice (glycation des intégrines) représentant le
signal modifiant certaines expressions géniques de la cellule endothéliale.
Ainsi des travaux de Charonis et Watanabe montrent quau niveau rétinien,
en présence d'une matrice de collagène glyqué ou de
laminine glyquée, la prolifération des péricytes est
réduite (diminution de la contractilité et hyperdébit)
alors que celle des cellules endothéliales est accentuée.
Conclusion sur les mécanismes biochimiques de la microangiopathie
:
Toutes ces perturbations biochimiques sont interdépendantes et
très liées entre-elles.
-
La voie des polyols : sorbitol entraîne une déplétion
en myoinositol mais donne aussi du fructose 7 à 8 fois plus glyquant
que le glucose.
-
La glycation de laldose réductase provoque son activation favorisant
la voie du sorbitol.
-
Laldose réductase consomme NADPH dont la déplétion
induit un déficit en piégeurs de radicaux libres et favorise
le stress oxydatif.
Des anomalies hémodynamiques précèderaient les modifications
structurales de la paroi vasculaire. En clair, les tissus réagissent
aux agressions dont ils sont victimes.
1/ Lésions
élémentaires
Les principales anomalies histologiques sont :
-
La raréfaction puis la disparition des péricytes.
-
La prolifération des cellules endothéliales dont les péricytes
et la membrane basale ne peuvent plus empêcher la croissance.
-
Lépaississement de la membrane basale (lipides, GAGS, fibronectine,
laminine et collagène IV) au fur et à mesure de lévolution
du diabète pouvant atteindre 5 fois lépaisseur normale.
Ces anomalies capillaires saccompagnent :
-
Dune hyperperméabilité, responsable dun dème avec
diffusion de lalbumine du plasma vers certains tissus et rupture des barrières
hémato-méningées et hémato-rétiniennes.
-
Dune diminution du tonus pariétal du à la perte des péricytes
entraînant la formation de micro-anévrismes.
-
Dune occlusion capillaire secondaire aux anomalies précédentes
et responsable dune ischémie tissulaire.
2/ Perturbations
hémo-rhéologiques
Fonctionnelles, contrairement aux lésions pariétales,
elles sont réversibles avec le retour à un parfait équilibre
glycémique.
-
Troubles hémodynamiques :
-
Augmentation du flux sanguin capillaire lors dune hyperglycémie
modérée (2 à 3 g/l) et au contraire diminution pour
une hyperglycémie sévère (> 4g/l).
-
Perte de lautorégulation du flux capillaire.
-
Conséquence : augmentation de la pression hydrostatique trans-capillaire
expliquant en partie lhyper-perméabilité de la paroi.
-
Diminution des résistances artériolaires, notamment celle
de lartère afférente glomérulaire qui diminue de
50% alors que la résistance de lartère efférente
reste inchangée. (ce qui entraîne une augmentation du flux
sanguin rénal et une albuminurie)
Tout ceci peut entraîner des lésions pariétales.
-
Troubles rhéologiques (augmentation de la viscosité sanguine
et plasmatique) dus à :
-
Une diminution de la déformabilité érythrocytaire,
gênante quand on sait quune hématie de 8m
m doit se déplacer dans un capillaire dont la lumière atteint
3 à 5m m .
-
Une augmentation de lagrégabilité érythrocytaire
corrélée à lélévation du fibrinogène.
-
Troubles de lhémostase :
Augmentation du fibrinogène et du fibrinopeptide
A (première étape de la transformation du fibrinogène
en fibrine sous laction de la thrombine) et diminution de la fibrinolyse
avec élévation du PAI 1 (plasminogène activateur inhibiteur
1 secrété par les cellules endothéliales)
Augmentation du facteur VII de coagulation (" colle
plaquettaire ")
Ces anomalies thrombogènes pourraient être secondaires
aux lésions micro-vasculaires ou corrélées à
léquilibre glycémique.
3/ CONSEQUENCES
: Hypoxie et Hypertrophie
-
Lhypoxie tissulaire entraîne un véritable cercle vicieux
puisquelle provoque une vasodilatation et une hyper-perméabilité
(par augmentation de pression) responsable ddème (fuite dalbumine)
majorant lhypoxie. De plus, elle aggrave les troubles rhéologiques.
Note : La photocoagulation au laser (fig. 14) permet de briser
ce cercle. En détruisant la rétine périphérique,
elle restaure la pression partielle en oxygène de la rétine
centrale restante. Loxygène pur permet alors une vasoconstriction
capillaire régulant le flux sanguin rétinien en le diminuant.
-
Lhypertrophie et lhyperplasie sont secondaires à lhypoxie
qui est un facteur de prolifération cellulaire, en particulier dangiogenèse
sous le contrôle de facteurs angiogéniques endogènes.
Ainsi comme nous lavons dit précédemment, les troubles hémorhéologiques
vont entraîner des modifications pariétales : prolifération
endothéliale. Mais ces modifications compensatrices aggravent le
processus car les cellules endothéliales vont dégénérer
entraînant la rupture des néovaisseaux et donc une hémorragie.
CONCLUSION : Actuellement, les chaînons qui relient
lhyperglycémie chronique aux lésions tissulaires restent
hypothétiques. Notons juste la nature pluri-factorielle de la microangiopathie.
Hormis lhyperglycémie, il convient de reconnaître limplication
de lHTA, des dyslipoprotéinémies, du tabac et de la grossesse
dans la survenue de cette pathologie. Ainsi lutter contre lapparition
de ce type de lésion revient à obtenir le meilleur contrôle
glycémique possible, mais aussi à rechercher et en traiter
toute élévation de la pression artérielle, à
lutter contre lexcès pondéral, le tabagisme et à
favoriser lactivité physique. Enfin, même si le diabète
de type II survient plus tard que le type I et laisse donc entrevoir une
chance plus minime dapparition dune microangiopathie, il est à
noter que DID et DNID comportent les même risques.

Figure 14
III Macroangiopathie et diabète
PHYSIOPATHOLOGIE
Elle désigne latteinte des artères musculaires et regroupe
deux maladies de la paroi artérielle : lathérosclérose
et une maladie dégénérative de la média, aboutissant
à la médiacalcose (sclérose artérielle caractérisée
par la dégénérescence et la calcification des fibres
musculaires de la média artérielle). Mais lathérosclérose
demeure la complication majeure puisque 50 à 75 % des diabétiques
insulino-dépendants meurent daccidents cardio-vasculaires. La prévalence
de lathérome est multipliée par deux chez les hommes diabétiques
et par quatre chez les femmes diabétiques, mais elle respecte la
répartition mondiale de lathérosclérose très
inégale dun pays à lautre. Ainsi le diabète najoute
pas un facteur de risque spécifique mais potentialise laction de
deux facteurs majeurs de risque vasculaire : lhypertension artérielle
(deux fois plus fréquente chez les diabétiques) et les dyslipoprotéinémies
(deux à trois fois plus fréquentes chez les diabétiques).
Le diabète ou plutôt les troubles hormonaux et métaboliques
ont des conséquences sur le métabolisme des lipoprotéines.
Dans le DID, ils sont secondaires au diabète et en lien avec léquilibre
glycémique. Alors que dans le DNID, ils sont secondaires à
linsulino-résistance, aggravés par le déséquilibre
qlycémique mais précèdent le diabète.
RAPPELS
-
Dans la circulation, les lipoprotéines lipases de la surface endothéliale,
activées par lApoprotéine C2, hydrolysent 4/5 des triglycérides
des chylomicrons.
-
En période de jeune, linsuline stimule la synthèse des lipoprotéines
lipases adipocytaires.
-
Le foie synthétise 90% des VLDL plasmatiques soit 30 à 50
grammes de TG par jour.
-
Les AG jouent un rôle important dans la régulation de la production
de lapo B100 : en stimulant la synthèse de lARNm de lapo B100
et en protégeant lapo B dun catabolisme précoce. Ainsi
une diminution de la synthèse de lapo B va entraîner une
diminution de la sécrétion de VLDL et une accumulation des
TG dans les hépatocytes entraînant une stéatose hépatique.
-
Les VLDL sont hydrolysés par les lipoprotéines lipases activées
par lapo C2 et dégradés en IDL puis pour la majeure partie
en LDL.
-
La concentration plasmatique des VLDL est la résultante de leur
synthèse hépatique et de leur catabolisme par les lipoprotéines
lipases.
-
Les LDL transportent 70 % du cholestérol.
-
70% des récepteurs des LDL sont situé au niveau du foie.
Ils sont internalisés avec la LDL, dégradée, libérant
du cholestérol libre dans le cytosol.
-
Les cellules assurent une autorégulation de leurs besoins en cholestérol.
-
Linsuline augmente le nombre de récepteurs B du foie.
Lorsque la voie du récepteur à lapo B est défaillante,
les
LDL saccumulent et vont soxyder au contact des cellules endothéliales.
Lapo B lysée nest plus reconnue par son récepteur. Néanmoins
les LDL oxydés ont une grande affinité pour le récepteur
" scavenger " des macrophages. Internalisées puis dégradées,
elles libèrent leur cholestérol qui est estérifié.
Mais les macrophages ne possèdent pas de rétrocontrôle
et se chargent de cholestérol se transformant en cellules spumeuses
qui semblent jouer un rôle fondamental dans la pathogénie
de lathérosclérose.
-
Les HDL sont synthétisées par le foie ou proviennent du catabolisme
des VLDL et des chylomicrons.
-
Elles captent du cholestérol libre à la surface des cellules
périphériques en se fixant par un récepteur de lapo
A1.
-
La LCAT estérifie le cholestérol.
-
Les HDL riches en cholestérol estérifié (HDL2) échangent
avec les VLDL, IDL et LDL du cholestérol estérifié
contre des TG grâce à la protéine de transport du cholestérol
estérifié CETP.
-
La lipase hépatique capte les TG et le cholestérol de ces
particules, régénérant des particules plus petites
et plus denses (HDL3).
-
Le cholestérol estérifié revient donc au niveau
du foie par lintermédiaire des HDL mais aussi grâce aux LDL
internalisés avec le récepteur à Apo B.
Le rôle de la protéine de transfert CETP dans la pathogénie
de lathérome reste débattu.
Une hypothèse serait que le transfert entraîne la
formation de LDL de petite taille, plus athérogènes que les
LDL synthétisés normalement par le foie.
La Lipoprotéine (a)
Dune structure voisine de la LDL, plus riche en triglycérides,
elle possède une apoprotéine spécifique : lApo (a)
reliée à lApo B par un ou plusieurs ponts dissulfures. Ces
liaisons masqueraient en partie le site de liaison de lApo B pour le récepteur
des LDL. Elle est synthétisée par le foie indépendamment
des autres lipoprotéines. Son taux est déterminé génétiquement
à 75%. Lathérosclérose a été associée
à des taux élevés de Lp(a). La mise en évidence
de la Lp(a) dans les plaques dathérome et la découverte
dun important degré dhomologie entre le plasminogène et
lApo (a) suggère que la Lp(a) pourrait constituer un lien étroit
entre les phénomènes dathérosclérose et de
thrombose.
-
Dyslipoprotéinémies dans le DID.
(fig. 15)
En cas dacidocétose, on observe une baisse du catabolisme des
chylomicrons et des VLDL dont les concentrations plasmatiques augmentent
entraînant une hypertriglycéridémie. En effet, la carence
insulinique induit un défaut dactivation de la lipoprotéine
lipase adipocytaire (insulino-sensible) qui normalement permet le stockage
desTG. Comme nous lavons rappelé, les HDL proviennent en partie
du catabolisme des VLDL et chylomicrons. Donc une baisse de ce catabolisme
rend compte de leffondrement des HDL. De plus, le catabolisme des LDL
par la voie des récepteurs Apo B du foie est ralenti pour deux raisons
: les LDL sont enrichis en TG du fait du défaut de délipidation
des VLDL et la carence en insuline provoque une diminution du nombre des
récepteurs (linsuline augmentant le nombre de récepteurs
Apo B du foie).
Linsulinothérapie corrige lensemble de ces troubles. Mais on
peut également observer des anomalies qualitatives des lipoprotéines
: augmentation du rapport cholestérol libre / lécithine (phospholipide
entrant dans la composition des lipoprotéines). Il y a altération
de la voie de retour du cholestérol des tissus vers le foie due
notamment à une augmentation du transfert du cholestérol
estérifié des HDL vers les VLDL et les LDL. Il sensuit une
diminution de lépuration tissulaire en cholestérol et une
augmentation de la production de LDL athérogènes.
En fait cest latteinte rénale secondaire au DID et la protéinurie
qui en résulte qui multiplie par 10 le risque de mortalité
cardiovasculaire. On note alors une élévation de la lipoprotéine
(a).
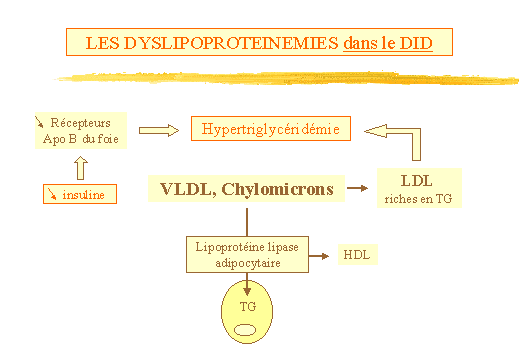
Figure 15
-
Dyslipoprotéinémies dans le DNID.
(fig. 16)
Un excès de synthèse des VLDL provoque une hypertriglycéridémie.
Cet excès de synthèse est secondaire à laugmentation
du flux portal de substrats : AG libres et Glucose. Lhyperinsulinisme
pourrait potentialiser laugmentation de synthèse des VLDL. En plus
de cette anomalie quantitative, on observe des anomalies qualitatives avec
des VLDL plus riches en triglycérides et cholestérol libre.
Leur taille est augmentée leur conférant une résistance
à la lipolyse. Certains vont être captés par les macrophages.
Linsulino-résistance caractéristique du Diabète
de type II avec surcharge pondérale rendrait compte de la moindre
activité de la lipoprotéine lipase adipocytaire.
Conséquences :
-
Lapport accru dacides gras libres et de Glucose au niveau du foie entraîne
une augmentation des VLDL et des IDL avec affinité
pour le récepteur Apo B du foie diminuée. Ces IDL sont athérogènes.
-
La lipolyse des VLDL est en diminution car ceux-ci y sont résistants.
Or le catabolisme des LDL est lui aussi en baisse (richesse en TG et diminution
des récepteurs due à linsulinopénie) ce qui crée
un équilibre entre anabolisme des LDL (lipolyse) et catabolisme
expliquant la stabilité relative du taux de LDL-cholestérol.
Mais les LDL sont de structure anormale, plus denses plus petits, riches
en TG et donc athérogènes.
Ainsi la voie de transformation des VLDL en LDL est diminuée
(la lipoprotéine lipase insulino-sensible voit son activité
réduite par linsulino-résistance) au profit de la production
de VLDL-remnants athérogènes.
-
On a des HDL anormales : - richesse en TG.
- capacité de transport du cholestérol diminuée.
-
Il y a diminution des HDL-cholestérol due à
lhyper-insulinisme secondaire à linsulino-résistance. En
effet, un taux anormalement élevé dinsuline entraîne
une hyper-activation de la lipase hépatique qui " transforme " HDL2
(riches en cholestérol estérifié) en HDL3.
Donc, lhypertriglycéridémie constitue un risque dathérogénèse.
Note : Mais lathérosclérose est aussi un marqueur
de risque dapparition dun DNID et pas
seulement la complication dun DNID.
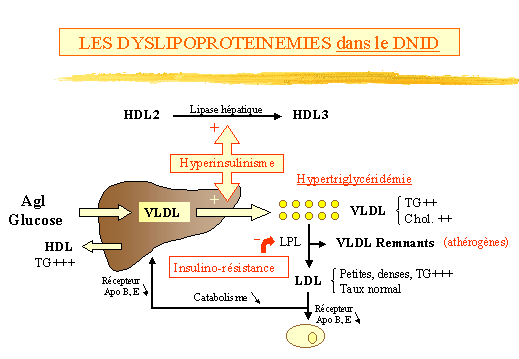
Figure 16
Le Syndrome X ou " pré-diabète " précède
le DNID.
Chez une personne obèse, dont la répartition du tissu
adipeux est de type androïde et viscérale, laugmentation de
la concentration en AGL est responsable dune insulino-résistance
entraînant une hyper-insulinémie qui associée à
des facteurs génétiques, à lage et des facteurs environnementaux
comme la sédentarité est susceptible dentraîner une
hypertension artérielle, un diabète non insulino-dépendant,
une hypertriglycéridémie (VLDL augmentés, HDL diminués)
et une hypofibrinolyse par diminution du plasminogène activateur
inhibiteur I (PAI I). (la plasmine lyse le caillot de fibrine)
-
Lhypertension artérielle chez le diabétique.
Environ 50% des diabétiques meurent daccidents cardio-vasculaires
suite à une athéromatose (Cf dyslipoprotéinémies)
et 30% des diabétiques insulino-dépendants décèdent
en insuffisance rénale terminale. Lhypertension artérielle
est un facteur déterminant de ces deux types de pathologies et son
traitement, sa surveillance notamment chez le diabétique vont être
essentiels à la prévention de lathérosclérose
et donc des macroangiopathies diabétiques.
-
LHTA ( > 140/90 mm Hg ) est plus fréquente chez le diabétique
mais de deux types suivant quil sagit dun diabète de type I ou
de type II. Elle a une prévalence denviron 15% chez les diabétiques
de type I et de 40% chez les diabétiques de type II. Dans le premier
cas, elle surtout secondaire à une glomérulopathie. En revanche,
dans le type II, même si ce cas de figure nest pas exclu, il sagit
plus souvent dune HTA essentielle associée à lobésité,
et qui, dans 50% des cas précède même le diabète.
Après 55 ans, ce sont 40% des hommes et 60% des femmes diabétiques
non insulino-dépendants qui sont hypertendus. Cest alors une HTA
systolique ou à prédominance systolique.
Les facteurs de risque sont : pour le diabète de type
I, la durée du diabète, léquilibre glycémique,
le sexe masculin, lexistence dune glomérulopathie ; pour le diabète
de type II, lâge, lindex pondéral, le sexe féminin
et lexistence dune atteinte rénale.
-
LHTA nest pas toujours consécutive au diabète. Elle
est secondaire dans la majorité des cas de diabète apparus
avant 30 ans. Mais, après 30 ans, lHTA précède une
fois sur deux lapparition dun DNID.
-
Rôle dans la formation de la plaque dathérome : les
facteurs de risque dapparition de maladies cardiovasculaires chez le diabétique
sont les mêmes que chez le non diabétique mais leur hiérarchie
est différente, avec un rôle majeur dévolu à
lhypertension artérielle systolique et à lhypertriglycéridémie.
-
Rôle dans lapparition de la cardiomyopathie diabétique
: lHTA serait responsable dune hypertrophie du septum et de la paroi
postérieure du ventricule gauche, avec un défaut de compliance
responsable dune gêne au remplissage ventriculaire.
Différentes formes dHTA du diabétique :
(Nous ne ferons ici que les évoquer brièvement)
-
Les hypertensions artérielles endocriniennes. Ex :
Hyperminéralocorticisme
-
Lhypertension artérielle réno-vasculaire, secondaire
à une sténose athéromateuse dune ou des deux artères
rénales et de leurs branches.
-
Lélévation tensionnelle de la néphropathie débutante.
-
Lhypertension artérielle de la glomérulopathie avec insuffisance
rénale (glomérulopathie patente). Lorsquil existe une
chute de la filtration glomérulaire, on assiste à une rétention
hydro-sodée avec augmentation des résistances périphériques
et une sensibilité accrue aux hormones pressives (noradrénaline,
vasopressine
) et donc la pression augmente ! Les diurétiques sont
donc logiquement le premier médicament anti-hypertenseur.
-
Lhypertension artérielle du diabétique âgé
ou ayant un vieux diabète. Il sagit dune hypertension artérielle
systolique ( PAS > 160mm Hg et PAD < 95mm Hg ). Le mécanisme
essentiel est un défaut de compliance des gros vaisseaux, perdant
leur fonction d'amortissement tensionnel et de régulateur de débit.
Cest le cas de la médiacalcose (macroangiopathie). La rigidité
artérielle augmente les forces de contrainte pariétales imposées
au cours du cycle cardiaque par les variations de pression artérielle.
Quand le débit augmente (systole) lélévation
de pression ne peut être amortie quavec une diminution des résistances
périphériques comme nous le montre la formule P=Q.R .
Or, le défaut de compliance empêche ce mécanisme
et donc entraîne une élévation de pression qui accélère
lusure des parois artérielles avec donc un véritable cercle
vicieux puisque celles-ci perdent encore de leur compliance. De plus lélévation
de la pression artérielle systolique est responsable dune augmentation
de la charge cardiaque susceptible dinduire une hypertrophie ventriculaire
gauche.
-
Lhypertension artérielle du diabétique non insulino-dépendant
obèse. Cest lexemple que nous allons étudier maintenant
au travers de sa physiopathologie :
Hypertension artérielle et obésité.
Cest le diabète de type II que nous retrouvons cette association
obésité et HTA. La parenté entre les deux maladies
est renforcée par lexistence de facteurs de risque communs : lâge,
le poids, la répartition androïde des graisses, la sédentarité.
La pathogénie de ce type dHTA est attribuée surtout à
linsulino-résistance et à lhyper-insulinisme qui en découle.
Linsulino-résistance entraîne une inhibition de la Na-K-ATPase
responsable dune rétention sodée intra-cellulaire, en particulier
au niveau des cellules musculaires lisses de la paroi vasculaire. Dautre
part, lhyper-insulinisme serait responsable dune rétention sodée
tubulaire, dune activation du système sympathique avec augmentation
du taux de noradrénaline, dune hypertrophie des cellules musculaires
lisses et peut-être dune activation du système rénine
angiotensine aldostérone. Ceci entraîne bien évidemment
une élévation de la pression artérielle. Le défaut
délimination sodée saccompagne dune volémie normale
puisque la surcharge sodée est essentiellement intra-cellulaire
(cellules musculaires lisses artériolaires). Cette rétention
entraîne une accumulation du calcium. La surcharge en sodium est
potassium intracellulaire serait responsable de lhypersensibilité
aux hormones vasopressives.
Les mesures diététiques et lactivité physique
ont une action parallèle sur la pression artérielle et linsulino-résistance.
En fait, comme nous lavons déjà vu, HTA et DNID sont
des manifestations du Syndrôme X.
Note : De fait, il existe une corrélation entre les valeurs
de pression artérielle systolique et diastolique, et, le rapport
périmètre de la taille sur périmètre des hanches
(répartition des graisses).
-
Deux profils diabétiques à haut
risque de macroangiopathie. (fig. 17)
-
Le diabétique non insulino-dépendant obèse, androïde
et sédentaire.
-
Le " diabétique néphropathe ".
-
Le premier cas comme nous lavons vu se caractérise par une insulino-résistance
et un hyper-insulinisme :
-
responsables de dyslipoprotéinémies : linsulino-résistance
entraîne une augmentation des substrats nécessaires au métabolisme
hépatique (AG libres et glucose), lhyper-insulinisme stimule la
synthèse des VLDL qui sont de structure anormale ( TG en augmentation
et diminution de lapo B ). De plus linsulino-résistance est responsable
dun défaut de catabolisme des VLDL par diminution dactivité
de la lipoprotéine lipase. Nous avons donc un profil athérogène
: VLDL et IDL élevés, HDL cholestérol en diminution.
-
Favorisant lhypertension artérielle : lhyper-insulinisme
comme nous lavons dit augmente le tonus sympathique (taux de noradrénaline
en hausse) et est responsable dune rétention sodée tubulaire,
tandis que linsulino-résistance diminue lactivité de la
Na-K-ATPase avec en conséquence une rétention sodée
et calcique intra-cellulaire, surtout au niveau des cellules musculaires
lisses de la paroi artérielle.
-
Plusieurs études comparant diabétiques insulino-dépendants
micro-albuminuriques aux diabétiques sans néphropathie incipiens,
ont montré que la micro-albuminurie saccompagne dune dyslipoprotéinémie
et dun profil thrombogène, si bien que la micro-albuminurie semble
être un témoin précoce dune vasculopathie généralisée.
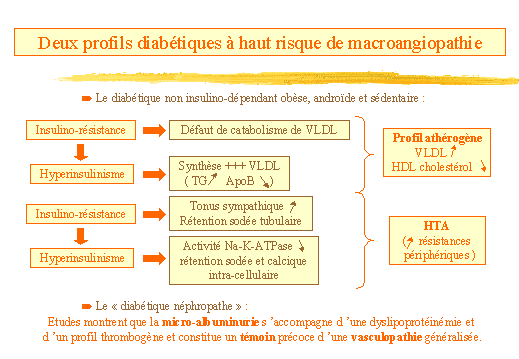
Figure 17
-
Athérosclérose diabétique.
Elle est plus précoce, plus fréquente et plus grave chez
les diabétiques quau sein dune population non diabétique.
Cette gravité accrue sexplique par laddition de plusieurs facteurs,
notamment la fréquence des troubles métaboliques ( par exemple,
lhypoglycémie favorise la thrombose), ou encore des altérations
vasculaires : microangiopathie diabétique, mais aussi artériolosclérose
avec épaississement de la média, et surtout médiacalcose,
conséquence dun vieillissement accéléré du
collagène artériel et/ou dun défaut dinnervation
pariétale secondaire à la neuropathie sympathique.
1/ Siége.
Elle touche essentiellement les artères coronaires, responsable
dinfarctus du myocarde, et les membres inférieurs ( artérite
des membres inférieurs ).
N.B. : au niveau cérébral, les lésions artériolaires
sont surtout dues à lhypertension artérielle.
2/ Rôle du diabète dans la pathogénie
de lathérosclérose. (fig. 18)
Rappelons brièvement les étapes de lathérosclérose
qui ne sont pas ici lobjet de notre exposé :
-
Elévation des LDL plasmatiques.
-
Infiltration des LDL dans lintima selon la pression artérielle
et la perméabilité de lendothélium.
-
Oxydation des LDL par des radicaux libres au contact des cellules endothéliales.
-
Les LDL sont reconnues par le " récepteur scavenger " des macrophages
et exercent leur pouvoir cytotoxique sur lendothélium dont elles
provoquent le nécrose et augmentent ainsi la perméabilité.
-
Transformation des macrophages en cellules spumeuses. Les macrophages sécrètent
entre autre des facteurs de croissance favorisant la prolifération
des cellules musculaires lisses capables de se transformer également
en cellules spumeuses par surcharge lipidique.
-
Formation de stries lipidiques (accumulation de cellules spumeuses dans
la couche sous-endothéliale).
-
Migration de cellules musculaires lisses de la média vers lintima,
sous laction de facteurs macrophagiques. Ces dernières proliférant
deviennent sécrétoires, synthétisant la chape fibreuse
entourant le cur athéromateux de la plaque.
-
Lésions endothéliales, néo-vascularisation de la plaque
avec des nécroses et des hémorragies.
-
Adhésion des plaquettes.
-
Libération de PDGF (facteur de croissance dorigine plaquettaire)
entraînant la migration et la prolifération des cellules musculaires
lisses.
-
Lésion évoluée, occlusion de lartère, ou rétrécissement,
ou détachage du thrombus et formation demboles.
Le diabète va intervenir à différents niveaux
(fig. 18) :
-
Lhyperglycémie entraîne des lésions endothéliales.
Elle induit une diminution de lhéparane sulfate qui provoque une
augmentation de la perméabilité endothéliale, favorise
la thrombose et stimule la prolifération des cellules musculaires
lisses. De plus lhyperglycémie chronique pourrait activer la protéine
kinase C induisant la synthèse de prostaglandines vasoconstrictrices
stimulant la prolifération des cellules musculaires lisses.
-
La glycation du collagène provoque un épaississement
pariétal et donc un défaut de compliance vasculaire, aggravant
les contraintes mécaniques de la paroi. Ce collagène glyqué
serait en outre un inducteur puissant de lagrégation plaquettaire.
Dautre part, les produits terminaux de glycation forment avec les LDL
des complexes qui se lient aux macrophages par des récepteurs spécifiques
et sont phagocytés. Enfin, lépaississement de la média
(médiacalcose) entraîne un ralentissement du flux des lipoprotéines,
augmentant leur temps de séjour dans lintima et favorisant donc
leur oxydation.
-
Comme nous lavons vu, le diabète entraîne une production
accrue de radicaux libres et une diminution des anti-oxydants endogènes,
favorisant loxydation des LDL.
-
Les macrophages captent :
-
Les LDL modifiés (oxydés et glyqués).
-
Les complexes antigènes (formes antigéniques de LDL) anticorps.
-
Les complexes LDL glycosaminoglycanes.
-
Les agrégats de LDL.
-
Les VLDL enrichis en TG (secondairement à lhyper-insulinisme).
-
Les IDL
-
Le diabète mal équilibré saccompagne dun profil
prothrombogène témoignant des anomalies fonctionnelles de
la cellule endothéliale (troubles de lhémostase déjà
envisagés dans la physiopathologie de la microangiopathie) :
-
diminution de la production de prostacycline anti-agrégante et accroissement
de la production de thromboxane A2 pro-agrégante.
-
augmentation de la production du facteur VIII (colle plaquettaire) et activation
du facteur VII.
-
augmentation du fibrinogène.
-
diminution de la fibrinolyse (lhyper-insulinisme induisant une augmentation
du PAI I)
-
Laugmentation de la viscosité sanguine (Cf troubles rhéologiques
dans la microangiopathie) favorise ladhérence des cellules sanguines
à lendothélium. Il sensuit une production accrue de facteurs
endothéliaux, notamment lendothéline qui est un puissant
vaso-constricteur et stimule la prolifération des cellules musculaires
lisses.
-
Linsuline pourrait stimuler la prolifération des cellules musculaires
lisses. Lhyper-insulinisme entraînerait un accroissement de lactivité
pro-proliférative des plaquettes qui est maximale chez les patients
à la fois hypertendus et diabétiques.
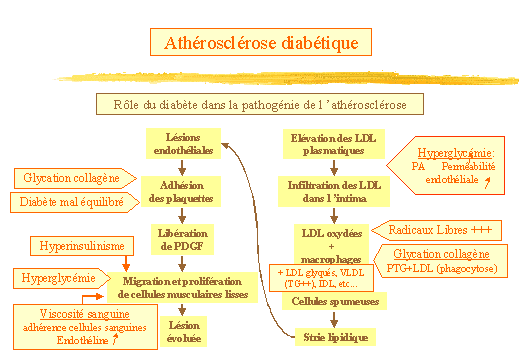
Figure 18
Latteinte cardiaque du diabétique consécutive à la
macroangiopathie est de deux types :
-
Il sagit surtout de lathérosclérose coronarienne : 50%
des diabétiques non insulino-dépendants et 25% des diabétiques
insulino-dépendants meurent dinsuffisance coronaire, cette ischémie
myocardique entraînant un infarctus du myocarde.
-
Mais on retrouve en plus des cardiomyopathies dues à :
-
Lhypertension artérielle.
-
La diminution de la compliance des gros vaisseaux.
CONCLUSION : Là encore, il apparaît clairement
que latteinte des gros vaisseaux ne peut à elle seule expliquer
la précocité, la fréquence et surtout la gravité
des atteintes périphériques comme ici la cardiopathie. En
effet, si lathérosclérose coronarienne est un facteur important,
la pathogénie ici évoquée est en fait pluri-factorielle
: la dénervation cardiaque (neuropathie) entraîne une modification
du rythme et une complication aiguë comme lhyperglycémie à
également des conséquences sur le myocarde en ischémie.
Retenons donc linteraction des différentes complications du diabète,
interaction qui transparaît de manière évidente dans
une complication bien connue du diabète : Le Pied diabétique.
IV Le Pied Diabétique
Plus de 50% des amputations de membre inférieur sont réalisées
chez des diabétiques.
On retrouve schématiquement 3 types de pied diabétique
:
-
Le pied neurologique (3/6)
-
Le pied artéritique (1/6)
-
Le pied mixte, cest à dire neurologique et vasculaire (2/6)
A cela vient sajouter linfection en cas dulcération.
Nayant pas étudié les Neuropathies, nous signalerons juste
que lon retrouve dans la pathogénie de la neuropathie diabétique
lensemble des processus physiopathologiques évoqués jusquici
: les angiopathies diabétiques se traduisant par des atteintes vasculaires
au niveau des vasa nervorum, par exemple une occlusion provoquant une ischémie
qui peut également être consécutive à un dème
lui-même secondaire à laugmentation de la perméabilité
capillaire (Cf voie des Polyols et Glycation protéique) et à
la rupture de la barrière hémato-nerveuse que lon retrouve
dans lhyperglycémie chronique. De plus, lhypoxie va être
à lorigine dune augmentation des radicaux libres alors quil y
a diminution des antioxydants endogènes. Enfin, noublions pas le
rôle de la déplétion en myoinositol dans laltération
des cellules nerveuses.
La neuropathie est donc responsable de diverses complications :
-
Lostéoarthropathie nerveuse (fig. 19), conséquence
dune dystrophie osseuse de dénervation et dun hyper-débit
sanguin avec ouverture de shunts artério-veineux. Ce dernier est
responsable dune male distribution sanguine avec dérivations du
flux sanguin vers les territoires cutanés et sous-cutanés
au détriment des territoires profonds (nerfs et os).
La marche impose au pied, en particulier ses points dappui des forces
de contraintes qui provoquent des fractures nécroses au niveau des
os fragilisés (ostéoporose) :
-
tête des métatarsiens
-
sommet de larche interne du pied (scaphoïde, 1er cunéiforme,
base du 1er métatarsien)
Ainsi, il y a effondrement de la voûte plantaire : le pied est plat
!
-
Le mal perforant (fig. 20) :
Les troubles moteurs sont responsables dune déformation progressive
du pied (pied creux et orteils en griffes), ce qui accentue lappui sous
la tête des métatarsiens, crée des frottements dans
la chaussure au niveau des articulations interphalangiennes proximales
et un appui amormal sur la pointe de lorteil.
Au niveau des points dappui et de frottements anormaux, se constituent
des durillons. Mais le diabétique neuropathe ne perçoit pas
la douleur et néglige ces durillons qui sont très durs et
blessent les tissus sous-cutanés. Une bourse séreuse se forme
sous le durillon. A la marche et à la station debout, le liquide
sous pression dissèque les tissus sous-cutanés qui se décollent.
Puis la couche cornée se fendille. La " bourse " nécrotique
sous-jacente se surinfecte. Un abcès se constitue dont le pus sévacue
quand la " coque " tombe.
-
Les blessures indolores par absence de perception de la douleur.
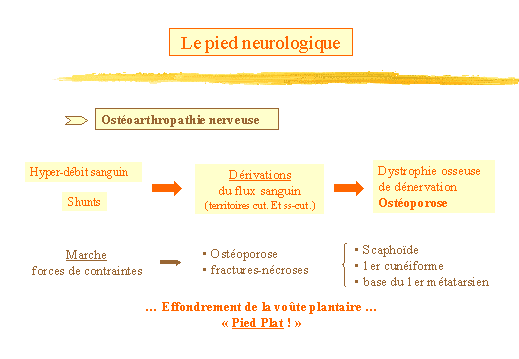
Figure 19
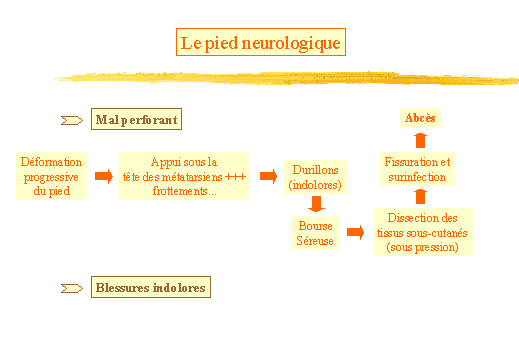
Figure 20
Rappels : Les lésions vasculaires des diabétiques
sont de 4 ordres :
-
Lathérosclérose.
-
Lartériolosclérose qui complique lhypertension artérielle
et participe à la perte de lautorégulation du flux capillaire.
-
La médiacalcose, secondaire à la dénervation sympathique
de lartère.
-
La microangiopathie qui intéresse les capillaires.
La gangrène sèche du diabétique est toujours la conséquence
dune athérosclérose des artères de gros et moyen
calibre. Les autres lésions vasculaires vont aggraver la gangrène.
Par exemple, la gangrène dun orteil nest pas secondaire à
la microangiopathie mais à des lésions dathérome
en aval et en amont avec une revascularisation par des vaisseaux collatéraux
au niveau de la cheville : doù des pouls pédieux et tibiaux
postérieurs perçus normalement.
Elle est rarement la cause du pied diabétique mais le complique
très fréquemment.
CONCLUSION GENERALE
La diminution du risque dapparition dune complication du diabète
(type I et type II) passe par la prévention. Ainsi, la fréquence
de survenue dun coma acido-cétosique est diminuée par léducation
du diabétique dont les éléments essentiels sont :
surveillance de la glycémie capillaire, de la glycosurie, adaptation
des doses dinsuline et contrôle de la cétonurie si nécessaire.
Enfin, nous avons vu que les facteurs de risque vasculaires sont multiples.
Il est important de souligner leur variabilité géographique.
Des études suggèrent l'importance des facteurs environnementaux,
principalement l'apparition dune alimentation de type occidental, dans
le développement de l'athérosclérose et des maladies
coronariennes chez les Japonais. En Europe, on observe de grandes variations
dans la prévalence des facteurs de risque chez des sujets coronariens.
Ces variations sont en partie imputables aux différences dans la
stratégie de prise en charge des sujets à risque coronarien
et dans les moyens en termes de spécialistes ou de financement mis
en oeuvre.
Sources :
- " Les Diabètes : comprendre pour traiter "
A. Grimaldi
-
Site de la Faculté de Médecine de Grenoble
http://www-sante.ujv-grenoble.fr/SANTE/
-
" Facteurs de risque Vasculaire "
H. Dabadie et H. Gin, Bordeaux.
- " La Biochimie " (Flammarion)
de Lubert Stryer
<Retour>
<Haut de la page>